2015/09/29
醫材申請臨床試驗是提升產品系統與公司品牌的價值 (Design Control: 確認未來與現在的需求)


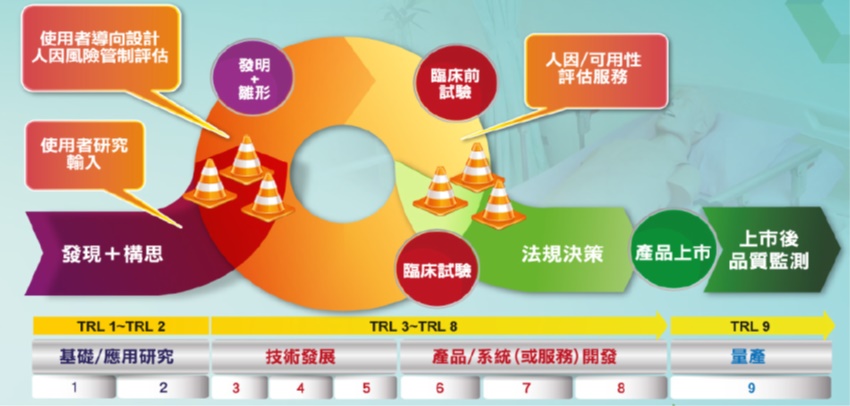
食藥署自102年起依據國內醫療器材產業發展趨勢,優先培育來自國內各大醫學中心相關科別(骨科、檢驗科、整形外科、心血管科、婦產科)之臨床試驗種子醫師,赴海外實地參與先進國家(美國、日本)之臨床試驗。期能經由早期介入高階醫療器材臨床試驗規劃及執行階段,通盤理解醫療器臨床試驗核心價值,並藉由相關產、學、研、醫界互動之過程,建立良好溝通橋樑,深化瞭解醫療器材臨床試驗產業上的創新價值,擴展國內醫療器材臨床試驗視野及強化跨國高階醫療器材研發合作量能,進而提升醫療器材產業全球化競爭力。
藉由建置國內優質醫療器材臨床試驗環境,促進國內高階醫材產品早日邁入「商品化」階段,提升國內臨床試驗品質與國際無縫接軌,加速國產新興醫療器材通過各國法規驗證,有效縮短上市前審查時程,預期將可提升醫材產值達35億元,並使國人及早使用安全有效之新興醫療器材。
臨床試驗案之臨床審查重點為何?
臨床試驗之臨床審查重點,涵蓋安全性評估、倫理考量、及試驗之科學意義等三大主軸,可區分成以下十點:
- 試驗之理論基礎及預期價值
- 受試者權益之維護
- 受試者安全之考量及維護
- 受試者之診斷
- 受試者納入、排除條件及試驗停止條件
- 劑量之選用、投與方式及治療期間
- 併用藥品之限制
- 療效指標之選擇
- 其他研究(PK/PD; PG Study)
- 受試者同意書
「試驗之理論基礎及預期價值」之審查重點為何?
臨床試驗必須有合理之試驗理論方可執行,試驗若尚處研發早期階段,試驗理論多依體外或動物試驗之藥理學、藥效學結果判定;倘若已有人體使用經驗,則亦應評估已知試驗結果是否足以支持本試驗之構想。試驗之價值則考量該試驗於研發過程中所扮演角色,及預期能提供何種資訊。
「受試者權益維護」之審查重點為何?
受試者權益之考量重點,在於判斷受試者是否會因參與試驗而喪失既有醫療權益。
- 若採安慰劑為對照組;
(1)須考量試驗期間內未接受積極治療,對安慰劑組病患之傷害性有多大?是否會造成不可回復之結 果,及其嚴重性如何?若有不可回復且嚴重傷害之疑慮,除非有「已無有效現行療法」、「非以安 慰劑為對照,試驗結果將不可信」等強烈理由,不宜採安慰劑為對照組。
(2)試驗期長短:為確保安慰劑組受試者之安全,宜選擇最短之試驗期,亦即試驗期間應在能達成試驗目的之合理範圍內儘量縮短。同時亦應注意試驗執行中,是否提供符合當時醫療水準之必要輔助醫療。
(3) 試驗結束之處置:盲性階段結束後,若能給予安慰劑組病患有效治療更佳。 - 若以有效藥品為對照組,應考量所選用之有效藥品是否為最適治療(optimal therapy),其用法用量是否合理。
- 為避免病患長期暴露於無效治療,損及病患權益,可考慮Add-on設計之可行性。但應同時考量Add-on設計對於療效及安全性評估之負面干擾。
「受試者安全之考量及維護」之審查重點為何?
試驗安全性考量之重點,在界定危險族群(risk group),並進而評估試驗風險是否獲得適當控制。
- 對於不需積極處置之短暫生理不適,因屬耐受性議題(tolerability issues),需確認是否符合告知同意要求。除既有人體使用經驗外,評估試驗藥品風險,亦會參考動物毒理試驗結果(target organ),及同機轉藥品之資訊(class effect)。
- 對於需積極醫療處置之安全性議題,需了解目前醫療處置中是否有可行之控制方法(例如增加檢查項目及密度)。若無適當控制方法,或試驗設計所採控制方法有不足之虞,需考慮是否列入排除條件,以排除危險除群。換言之,安全性評估之步驟須先界定高危險族群,並評估保 護措施是否適當,而保護措施以適當控制風險為先,不得已時才採排除手段。
「受試者納入、排除條件及試驗停止條件」之審查重點為何?
納入及排除條件之評估重點,著重於受試者安全性之維護。 請依照「3.受試者安全之考量及維護」之重點,審慎界定危險族群。
- 臨床試驗通常會就受試者基本器官功能及健康狀況,訂有最低要求,例如肝臟、腎臟、心臟、肺臟、Eastern Cooperative Oncology Group (ECOG) score、Karnofsky Performance Scale (KPS)等相關規定。審查時將依照試驗風險,考量其所定標準之合理性。例如,已知可能影響肝功能之藥品,需特別注意納入排除條件中對於基礎肝功能之限制。
- 對於高危險族群之保護措施,優先考量風險控制方法,若無法控制受試者參加試驗之危險性, 才列為排除(或限制)條件。
- 審查受試者納入、排除條件時,會兼顧試驗之可執行性,及所擇取群體之代表性。
「受試者同意書」之審查重點為何?
- 受試者同意書內容之評估,原則上儘量尊重人體試驗委員會(IRB)之決定及建議。
- 藥物基因學研究相關之受試者同意書宜參考藥政處於94年10月13日(衛署藥字0940338555號)公告之「藥物基因體學研究之受檢者同意書內容參考指引」。
臨床試驗案於何種情況下,需請廠商補件提供資料(臨床部分)?
- 有損害受試者權益之虞者。
- 有不合理之安全性疑慮者。
- 所提供之臨床前或臨床試驗資料有嚴重瑕疵,或明顯違反醫學知識,導致無法支持試驗可行性者。
- 試驗設計方面之議題,以違反科學原則,導致試驗目的顯然無法達成者為限。